
На Республиканской научно-практической конференции «Актуальные вопросы пульмонологии и фтизиатрии», состоявшейся в рамках 31-го Международного медицинского форума «Здравоохранение Беларуси — 2026», старший преподаватель кафедры кардиологии и внутренних болезней БГМУ Татьяна Войтко представила доклад на тему «Редкие заболевания в пульмонологии — что мы видим сегодня».
Частота и факторы
Редкими считаются заболевания, частота которых составляет менее одного случая на каждые 2 000 человек (в Европе), то есть это орфанные заболевания — малоисследованные состояния, заболевания, специфические методы лечения которых пока не разработаны. Около 80 % редких заболеваний обусловлено генетическими факторами (см. таблицу), также они могут быть легочными проявлениями системного заболевания либо вызваны каким-либо лекарственным препаратом, предназначенным для лечения другого заболевания.
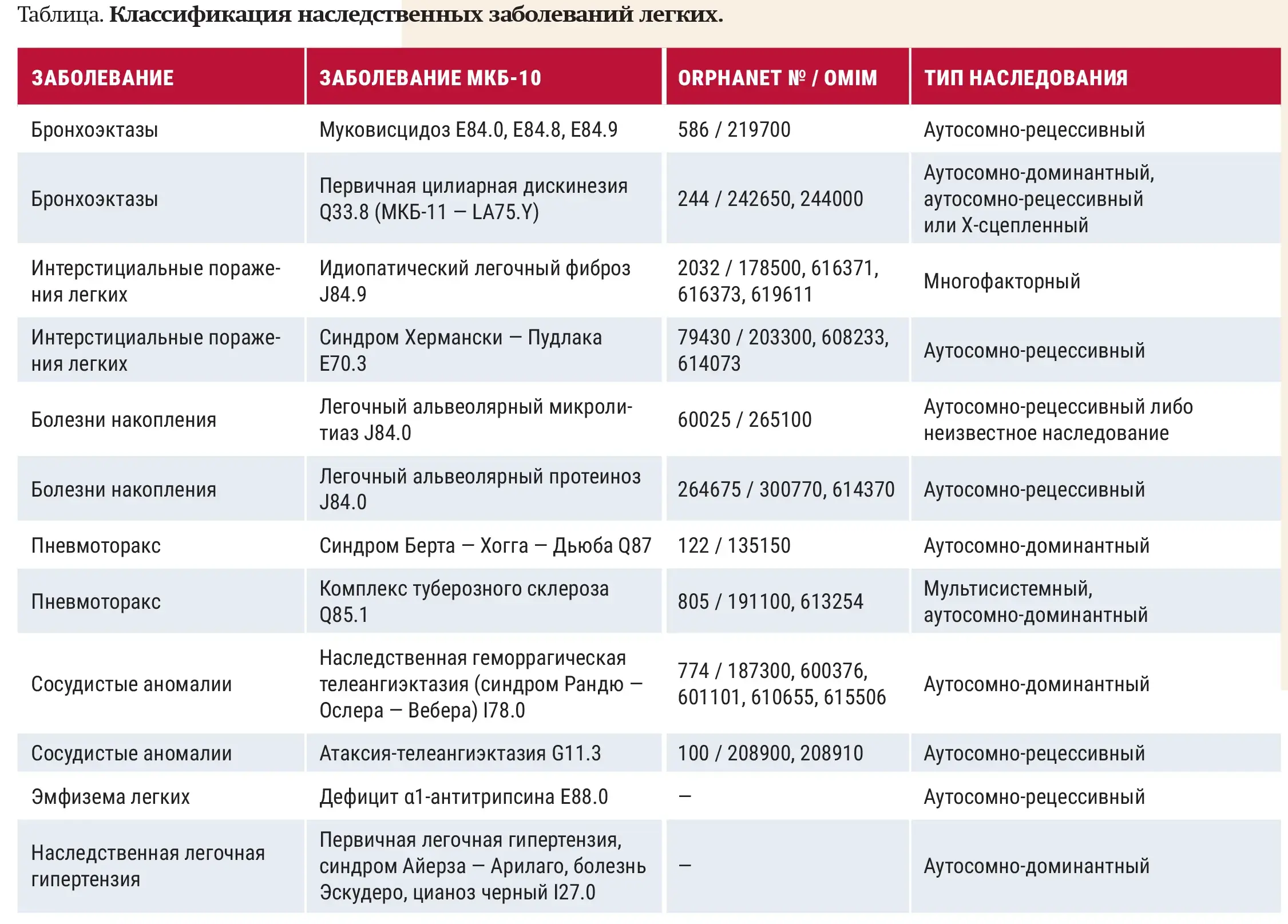
Основные современные тенденции сосредоточены на углублении генетической диагностики, выявлении общих механизмов старения и переходе к персонализированной медицине.
 Татьяна ВойткоТак, много внимания уделяется генетической архитектуре и новым локусам, ведь научные исследования значительно расширили понимание генетической основы заболеваний легких.
Татьяна ВойткоТак, много внимания уделяется генетической архитектуре и новым локусам, ведь научные исследования значительно расширили понимание генетической основы заболеваний легких.
Основные достижения включают идентификацию специфических генов при идиопатическом легочном фиброзе (ИЛФ): у взрослых основными генетическими причинами фиброза легких являются мутации в генах, связанных с теломерами (например, TERT, TERC, RTEL1); в детском возрасте доминируют мутации в генах, связанных с сурфактантом (SFTPB, SFTPC, ABCA3). Все большее значение приобретает оценка совокупного генетического риска (Polygenic Risk Scores, PRS), которая позволяет предсказывать вероятность развития болезней даже при отсутствии одной сильной мутации.
Одной из самых ярких тенденций стало обнаружение общих генетических факторов для разных заболеваний легких. Это, во-первых, общие локусы: гены FAM13A, DSP и TERT связаны одновременно с ИЛФ, хронической обструктивной болезнью легких (ХОБЛ) и раком легких.
Во-вторых, эффект противоположных аллелей: интересно, что одна и та же аллель может повышать риск ИЛФ, но при этом снижать риск развития ХОБЛ или рака легких, что указывает на существование биологических компромиссов в стареющем легком.
Появляются центры, ориентированные именно на семейные формы интерстициальных заболеваний легких (FIP clinics), где проводится скрининг родственников первой степени. Современные рекомендации включают ежегодное проведение КТ высокого разрешения и функциональных легочных тестов для бессимптомных родственников пациентов с семейным фиброзом. Еще одна тенденция — таргетная терапия. Успех применения ингибиторов mTOR при лимфангио-лейомиоматозе (ЛАМ) стал доказательством эффективности терапии, направленной на конкретный генетический дефект.
Предлагаются такие меры, как неонатальный скрининг, поскольку расширение программ скрининга новорожденных позволяет выявлять такие болезни, как муковисцидоз (МВ), первичная цилиарная дискинезия (ПЦД) в первые дни жизни, значительно улучшая прогноз; создание национальных и международных регистров пациентов (например, по МВ, по ПЦД) для сбора данных, проведения клинических исследований и стандартизации лечения; радиологические методы (использование низкодозной КТ для безопасного мониторинга состояния легочной ткани, особенно у детей).
Муковисцидоз (МВ)
Муковисцидоз, кистозный фиброз — тяжелое наследственное заболевание, при котором поражаются железы внешней секреции, в результате чего они вырабатывают слишком густую слизь. Частота распространения в Беларуси составляет 1:6 000 новорожденных. Частота носительства — 1:40–45 (без клинических проявлений). При этом аутосомно-рецессивном заболевании нарушены или отсутствуют функции белка CFTR, что вызывает каскад нарушений во всем организме: продукция густой слизи → присоединение инфекции → воспаление → слизь становится еще более вязкой → возникает порочный круг.
Таким образом, МВ — это прогрессирующее полиорганное заболевание, основные клинические проявления которого связаны с поражением дыхательной системы (рецидивирующие легочные инфекции со стойким кашлем и густой, гнойной, трудноотделяемой мокротой, формированием бронхоэктазов, пневмофиброза, ателектазов, одышкой) и пищеварительной системы (панкреатическая недостаточность), характеризующееся снижением массы тела (иногда роста). У детей грудного возраста отмечается соленый вкус кожи, боли и спазмы в животе, у новорожденных возможно развитие мекониальной кишечной непроходимости. У взрослых пациентов отмечается снижение фертильности. Снижение функции CFTR связано с тяжелыми клиническими проявлениями заболевания и высоким риском смертности. Активность этого белка — определяющий фактор клинических исходов у пациентов с МВ: чем выше активность CFTR, тем менее выражены клинические проявления и качество жизни пациентов выше.
Благодаря совершенствованию диагностики и лечения МВ во всем мире наблюдается неуклонный рост средней продолжительности жизни пациентов. МВ трансформировался из фатального заболевания детства в хроническую болезнь взрослых.
В Беларуси число взрослых пациентов с МВ увеличилось за последние 20 лет более чем в 9 раз (с 9 пациентов в 2005 году до 85 в 2025-м), медиана возраста достигла 36 лет (18–50 лет), что говорит об улучшении диагностики и лечения. Значительно улучшилось качество жизни пациентов, выявлены особенности течения заболевания, его генетические аспекты.
Принят ряд документов и постановлений по диагностике и лечению этого и других генетических заболеваний. Диагностика МВ включает клинические проявления, проведение потового теста, генетическое обследование (медико-генетическое консультирование).
Специализированную помощь пациентам оказывают как в Минске (БГМУ, МНПЦ хирургии, трансплантологии и гематологии, Минский клинический консультативно-диагностический центр), так и в областных центрах (Гродненская, Минская, Витебская, Брестская ОКБ), в РНПЦ пульмонологии и фтизиатрии. Лечение включает медикаментозную коррекцию (заместительная ферментная терапия мультиэнзимными препаратами, жирорастворимые витамины, муколитики, бронходилататоры), а также методы физиокинезитерапии.
Пациенты имеют право на льготное (бесплатное) лекарственное обеспечение, которое включает заместительную ферментную терапию препаратами Креон 10 000 и 25 000, ингаляционную терапию (КДБА, ДДБА, ИГКС, М-холинолитические препараты), системные глюкокортикостероидные препараты (по показаниям), колистин для ингаляций, тигеразу. Продолжается оптимизация программ лечения и реабилитации, предполагается расширение доступа к инновационным методам терапии (использование таргетных препаратов), улучшение диагностики и развитие трансплантационной помощи.
Первичная цилиарная дискинезия (ПЦД)
Это второе по частоте наследственное заболевание дыхательных путей после муковисцидоза. ПЦД (цилиопатия) — генетическое заболевание, передающееся по аутосомно-рецессивному типу (обусловлено мутациями в генах, отвечающих за структуру и функцию ресничек (цилий), выстилающих дыхательные пути, пазухи носа и внутреннее ухо).
С заболеванием связано более 40 различных генов, из которых наиболее распространенные — DNAH5, DNAI1 и RSPH4A, описаны и более редкие Х-сцепленные формы. Известного аутоиммунного компонента ПЦД не существует. Частота встречаемости заболевания — примерно 1/15 000–1/30 000 новорожденных, однако, по некоторым оценкам, данные занижены и заболевание встречается чаще. ПЦД поражает в равной степени как мужчин, так и женщин.
Общие клинические симптомы ПЦД включают хронический кашель, который может сопровождаться выделением мокроты; частые ОРИ, бронхиты, пневмонии, нередко с затяжным течением; формирование бронхоэктазов; синусит (риносинусопатии); рецидивирующие отиты. Кроме того, у мужчин наблюдается снижение подвижности сперматозоидов, а у женщин могут возникнуть проблемы с транспортировкой яйцеклеток.
Диагностика включает клинические проявления, рентгенографию грудной клетки или компьютерную томографию (для оценки структуры легких и выявления осложнений, таких как бронхоэктазы), назальный тест на оксид азота (у людей с ПЦД часто бывает ниже); на цилиарную функцию (оценка движения ресничек в образце, взятом из слизистой оболочки носа).
Генетическое тестирование направлено на выявление мутаций в известных генах, связанных с ПЦД.
Заболевание обычно диагностируется в детстве, но симптомы могут проявиться и в более позднем возрасте. Риску ПЦД (ключевые факторы) подвержены лица, в семейном анамнезе которых есть случаи респираторных заболеваний, нередко в сочетании с врожденными пороками сердца.
Лечение симптоматическое: муколитики, бронходилататоры, антибактериальные препараты (с учетом чувствительности микрофлоры мокроты). В некоторых случаях для устранения осложнений (хронический синусит или инфекции уха) может потребоваться хирургическое вмешательство. Немедикаментозное лечение включает методы очистки дыхательных путей (ингаляционная и физиокинезитерапия), диету (сбалансированный рацион, богатый антиоксидантами и противовоспалительными продуктами), модификацию образа жизни (отказ от курения и минимизация воздействия раздражителей дыхательных путей).
Интерстициальные патологии
Распространенность идиопатического легочного фиброза (обычная интерстициальная пневмония по гистологической классификации) широко варьирует, частично из-за регионов с более строгим скринингом, при этом ежегодно в США некоторые оценки показывают заболеваемость 495 на 100 тысяч человек.
Существует некоторая генетическая предрасположенность (в особенности полиморфизмы гена MUC5B, которые вызывают гиперэкспрессию муцина), наблюдается примерно в 30–35 % случаев.
Основной фактор, коррелирующий с заболеванием, — курение, как в настоящем, так и в прошлом. Клинические симптомы: сначала появляются сухой кашель и одышка при физической нагрузке, затем одышка становится постоянной, отмечается снижение массы тела и изменение пальцев по типу «барабанных палочек».
Основа диагностики — КТ высокого разрешения, которая позволяет выявить «сотовое легкое», морфологическое исследование при проведении торакобиопсии. Диагноз чаще всего ставится пациентам в возрасте 60–70 лет, лечение симптоматическое, в тяжелых случаях — трансплантация легких.
Современные тренды предлагают молекулярно-генетическую диагностику — широкое внедрение секвенирования нового поколения (NGS), особенно при редких интерстициальных заболеваниях легких у детей, а также генную терапию и коррекцию — использование липидных наночастиц для доставки мРНК в клетки легких, что позволяет восстанавливать функцию дефектных генов. Таргетная терапия направлена на разработку и применение препаратов, воздействующих на молекулярную причину болезни, а не только на симптомы.
Легочный альвеолярный протеиноз (ЛАП)
Заболевание обусловлено нарушением клиренса или продукции сурфактанта, альвеолы заполняются бесклеточным липопротеиновым ШИК-положительным сурфактантом, при этом клетки альвеол и интерстиция без патологии. Поражаются преимущественно заднебазальные сегменты легкого, а плевра и средостение не вовлекаются. Распространенность в популяции оценивается примерно 7–10 случаев на миллион.
ЛАП чаще всего идиопатический и встречается у здоровых людей в возрасте 30–50 лет. Считается, что заболевание чаще поражает мужчин и связано с курением.
Редкие вторичные формы встречаются у пациентов с силикозом, при проведении иммуносупрессивной терапии, у онкогематологических больных, при ингаляционном воздействии алюминиевой, титановой, цементной и целлюлозной пыли и др.
Заболевание проявляется одышкой, недомоганием, усталостью. Заподозрить ЛАП можно при выявлении на рентгенограмме органов грудной клетки двустороннего затенения в средних и нижних отделах легких в виде бабочки с нормальными корнями легких. Для диагностики используются лабораторные исследования, проведение бронхоальвеолярного лаважа, иногда биопсии легких. Лечение предполагает бронхоальвеолярный лаваж, в некоторых случаях назначение рекомбинантного гранулоцитарного колониестимулирующего фактора макрофагов.
На фоне лечения 5-летняя выживаемость составляет около 80 %. Трансплантация проводится нечасто, так как заболевание развивается и в пересаженном легком.
Лимфангиолейомиоматоз (ЛАМ)
Заболевание редкое, встречается только у женщин, как правило, в возрасте от 20 до 40 лет, наибольшему риску подвержены белые женщины. ЛАМ поражает <1 из миллиона человек в год; однако с развитием скрининга распространенность увеличивается (2 случая на каждые 10 000). Характеризуется пролиферацией атипичных гладкомышечных клеток повсеместно в грудной клетке, включая легочную паренхиму, сосудистую, лимфатическую системы и плевру, что приводит к искажению архитектуры легких, кистозной эмфиземе и прогрессирующему ухудшению функции легких. Может возникать у пациентов с туберозным склерозом (ТС), ТТС-ЛАМ, или как спорадический ЛАМ (у пациентов без ТС). Патогенез ЛАМ включает пролиферацию, лимфогенез, лимфатическое распространение, имплантацию, деструкцию тканей.
Существуют разные варианты течения ЛАМ, но обобщенная клиническая картина заболевания представлена на рисунке.
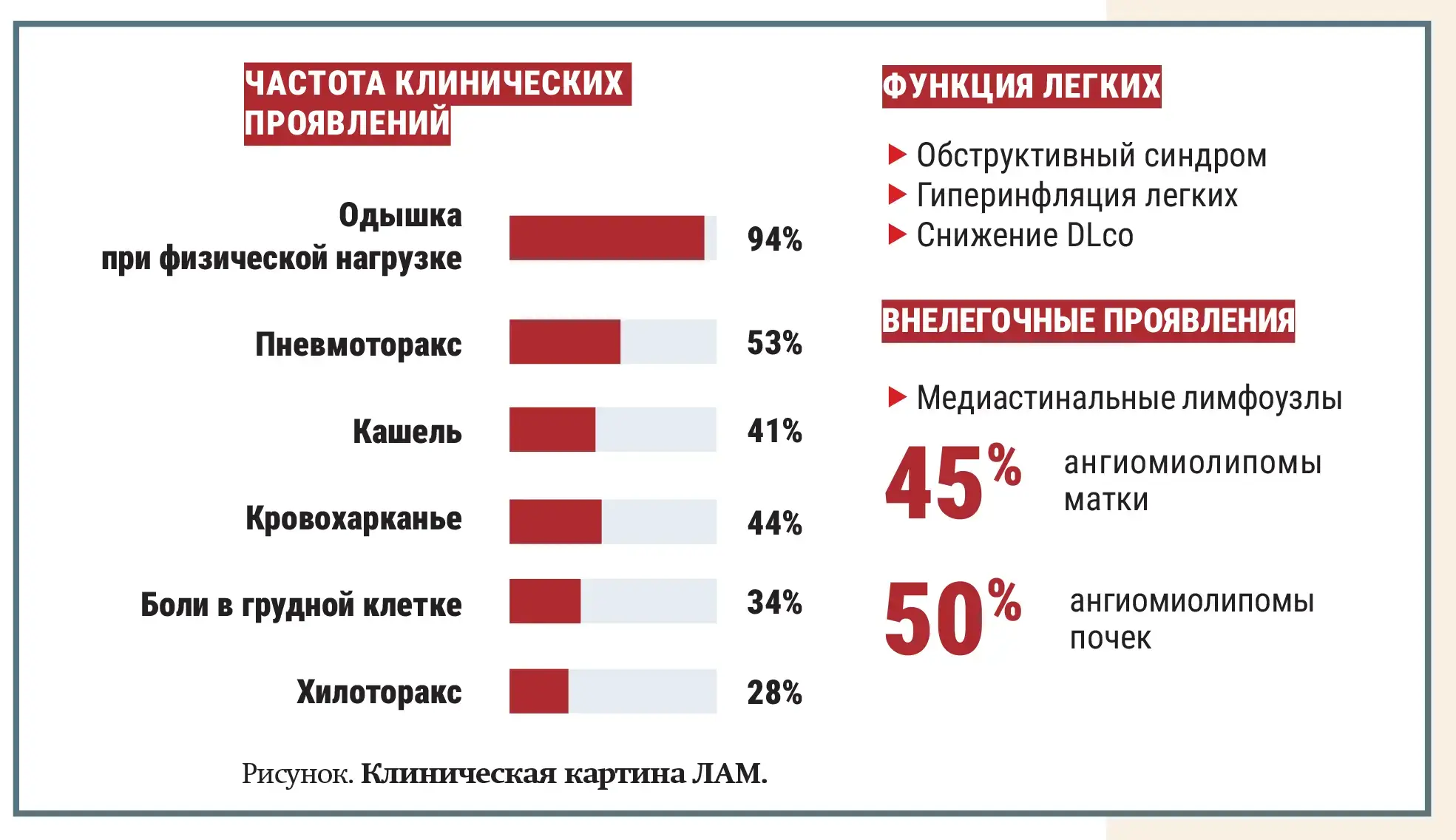
Диагностика включает: компьютерную томографию с отображением кист легкого; анализ хилезного выпота (при туберозном склерозе); анализ крови, показывающий высокий уровень протеина — фактора роста эндотелия сосудов D (VEGF-D); в некоторых случаях может потребоваться выполнение биопсии легких.
У 50 % пациентов с ЛАМ выявляется ангиомиолипома — доброкачественная опухоль почек, с риском кровотечения, если она больше 4 см.
Для лечения предлагаются кислородотерапия, плевродез или плеврэктомия (для спавшегося легкого). Из медикаментов — сиролимус (эверолимус, рапамицин), который помогает остановить снижение функции легких (но требуется не всем). Контроль симптомов: поддержание нормального веса; отказ от курения; активный образ жизни; программа легочной реабилитации для борьбы с одышкой; использование ингаляторов при синдроме бронхиальной обструкции; вакцинация от гриппа и пневмококковой инфекции. Важны также отказ от комбинированных оральных контрацептивов (содержащих эстроген и прогестерон) и обсуждение беременности; отказ от заместительной гормональной терапии после наступления менопаузы. При дальнейшем снижении легочной функции и прогрессировании дыхательной недостаточности показано проведение трансплантации легких.
Пульмональный гистиоцитоз Лангерганса (ПГЛ)
Заболевание, при котором соматически мутировавшие моноклональные CD1a-положительные клетки, подобные клеткам Лангерганса (тип гистиоцитов или антигенпрезентирующих клеток легких), инфильтрируют бронхиолы и альвеолярный интерстиций вместе с лимфоцитами, плазматическими клетками, нейтрофилами и эозинофилами. Как правило, поражает средние и верхние доли легкого.
Типичные клинические проявления: одышка, непродуктивный кашель, утомляемость, потеря веса, плевритическая боль. Часто встречается внезапный, спонтанный пневмоторакс. Примерно у 15 % пациентов с ПГЛ симптомы отсутствуют.
КТ высокого разрешения показывает характерное наличие кист в средней и верхних долях (часто причудливой формы) и/или очаговых образований с утолщением интерстиция). Для диагностики важно исследование функции легких, в некоторых случаях — бронхоскопия и биопсия. Причина смерти — дыхательная недостаточность или рак.
Лечение предполагает отказ от курения, эмпирическое применение глюкокортикоидов и цитотоксических препаратов (кладрибина, цитарабина, винбластина), однако данных крупных клинических исследований недостаточно, а также трансплантацию легких при прогрессирующей дыхательной недостаточности (заболевание может рецидивировать в пересаженном легком, если пациент продолжает курить).
Спонтанное исчезновение симптомов встречается у некоторых пациентов с минимальными проявлениями лангергансоклеточного гистиоцитоза. Десятилетняя выживаемость составляет >90 %.
Дефицит альфа-1-антитрипсина
Генетическое заболевание, характеризующееся нехваткой ингибитора легочных протеаз альфа-1-антитрипсина, которая приводит к усиленной, обусловленной протеазами деструкции тканей легких и развитию эмфиземы у взрослых. Накопление в печени патологического альфа-1-антитрипсина может привести к развитию болезни печени как у детей, так и у взрослых. Встречается в среднем с частотой 1:3 000–5 000 в популяции, при этом для балтийского региона и прилегающих областей (включая Беларусь) характерна одна из самых высоких частот генотипа PiZZ (1:1 500–1:2 000).
Заболевание часто остается недодиагностированным.
Недостаточность альфа-1-антитрипсина можно заподозрить:
- у курильщиков (развитие эмфиземы в возрасте до 45 лет);
- людей, которые не курят и не подвергаются производственному воздействию (эмфизема развивается в любом возрасте);
- пациентов, у которых выявляют преимущественное распределение эмфиземы в нижних долях легких;
- пациентов с семейным анамнезом эмфиземы или цирроза без четко выявленной причины;
- людей с семейным анамнезом дефицита альфа-1-антитрипсина;
- пациентов с панникулитом; новорожденных с желтухой или повышением ферментов печени;
- пациентов с бронхоэктазами неясной этиологии или заболеванием печени.
Патогенез и клинические проявления в легких: дефицит альфа-1-антитрипсина способствует разрушению тканей, приводящему к эмфиземе (особенно у курящих), причем поражение легких редко встречается у пациентов моложе 25 лет; возможно формирование бронхоэктазов. В печени: изменение конформации молекулы альфа-1-антитрипсина вызывает полимеризацию и накопление аберрантных молекул внутри гепатоцитов, вызывая холестаз, а позднее — формирование цирроза (1/3 пациентов) и повышает риск развития гепатоцеллюлярной карциномы. Другие проявления: панникулит, кровотечения, язвенные колиты, АНЦА-ассоциированный васкулит с поражением клубочков почек.
Диагностика проводится с помощью генотипирования и подтверждается при сывороточных уровнях альфа-1-антитрипсина <80 мг/дл (<15 мкмоль/л). В Беларуси проводятся биохимические и молекулярно-генетическое исследования гена SERPINA1 для выявления мутаций (PiS- и PiZ-фенотипы).
При заболеваниях легких часто назначают заместительную терапию очищенным человеческим альфа-1-антитрипсином. Кроме того, обязателен отказ от курения, в некоторых случаях прибегают к трансплантации легкого или печени. Лечение панникулита симптоматическое (назначаются дапсон, кортикостероиды и тетрациклины).
Синдром Берта —Хогга — Дьюба
Редкое наследственное аутосомно-доминантное заболевание, вызванное мутацией в гене-супрессоре опухолей FLCN (хромосома 17p11.2), кодирующем белок фолликулин (в мире описано не менее 600 случаев).
Характеризуется триадой симптомов: доброкачественными опухолями кожи (фиброфолликуломы), кистами легких с риском пневмоторакса и повышенным риском развития почечно-клеточного рака.
Диагностика основывается на клинических проявлениях, отягощенном семейном анамнезе, молекулярно-генетическом анализе, дерматологическом осмотре, КТ легких и МРТ/КТ почек. Синдром не имеет собственного отдельного кода в МКБ-10, его часто относят к рубрике врожденных пороков развития (например, Q87.8).
Кожные поражения чаще появляются в 3–4-м десятилетии жизни в виде мелких папулезных высыпаний (фиброфолликуломы, триходискомы) на лице, шее и верхней части груди. Легочные симптомы: образование кист в легких, которые часто приводят к спонтанному пневмотораксу. Почечные опухоли: высокий риск развития различных видов рака почек, часто двусторонних и многоочаговых.
Специфического лечения генетического дефекта нет. Терапия направлена на контроль симптомов: лазерная терапия или хирургическое удаление кожных образований; наблюдение и при необходимости — лечение пневмоторакса; регулярный скрининг почек (МРТ/УЗИ). Пациентам с данным синдромом крайне важно пожизненно наблюдаться у дерматолога, уролога и пульмонолога.