
Опухоли центральной нервной системы (ЦНС) у пациентов детского возраста занимают второе место среди всех онкологических заболеваний и первое среди солидных новообразований, являются второй по частоте причиной смерти от злокачественной патологии. Стандартизованный показатель заболеваемости (мировой стандарт) данной патологией в Беларуси за 27-летний период (1997–2024) составил 3,12 на 100 тысяч детского населения, что соответствует среднему уровню заболеваемости детей новообразованиями ЦНС в странах Европы.
Сегодня основные усилия направлены на поиск максимально возможной «расшифровки» генетического опухолевого кода с его точной молекулярной характеристикой. Молекулярный фенотип опухоли может быть эффективно использован для более точного прогнозирования клинического исхода заболевания, посттерапевтического мониторинга качества ремиссии, а также для лечения, подобранного под индивидуальный опухолевый профиль конкретного пациента. В РНПЦ ДОГИ накоплен опыт применения современных методов ведения детей с опухолями ЦНС.
Морфологические варианты первичных глиом
 Анжелика СолнцеваПервичные опухоли ЦНС нейроэпителиального происхождения (глиомы) составляют 75 %. Низкозлокачественные глиомы 1-й и 2-й степени злокачественности диагностируют в 41,7 % случаев, на высокозлокачественные глиомы у детей приходится 15–20 %.
Анжелика СолнцеваПервичные опухоли ЦНС нейроэпителиального происхождения (глиомы) составляют 75 %. Низкозлокачественные глиомы 1-й и 2-й степени злокачественности диагностируют в 41,7 % случаев, на высокозлокачественные глиомы у детей приходится 15–20 %.
Показатели 10-летней общей выживаемости (ОВ) и выживаемости без прогрессии (ВБП) у детей с низкозлокачественными глиомами (НЗГ) в Беларуси составляют 81 % и 70 % соответственно. У пациентов с высокозлокачественными глиомами (ВЗГ) показатели 5-летней ОВ и ВБП — 25 % и 15 %.
В актуальной классификации опухолей ЦНС Всемирной организации здравоохранения 2021 года, кроме традиционных морфологических характеристик опухоли, для постановки окончательного диагноза обязательно требуется определение молекулярно-генетических характеристик новообразования.
Морфологические варианты НЗГ представлены пилоцитарными астроцитомами, дисэмбриопластическими нейро-эпителиальными опухолями и диффузными низкозлокачественными  Леонид Киселевглиомами педиатрического типа. НЗГ возникают как в рамках наследственного синдрома — нейрофиброматоза 1-го типа (НФ1), так и спорадически (характеризуются более агрессивным течением и разнообразным профилем молекулярно-генетических аберраций). В случае радикальной резекции высока вероятность полного излечения без какой-либо дополнительной терапии.
Леонид Киселевглиомами педиатрического типа. НЗГ возникают как в рамках наследственного синдрома — нейрофиброматоза 1-го типа (НФ1), так и спорадически (характеризуются более агрессивным течением и разнообразным профилем молекулярно-генетических аберраций). В случае радикальной резекции высока вероятность полного излечения без какой-либо дополнительной терапии.
Главную проблему при опухолях ЦНС представляют случаи с локализациями новообразования в срединных структурах мозга: хиазмально-селлярной области, стволе, таламусе и др.
Радикальное удаление опухоли с таким расположением невозможно вследствие несовместимых с жизнью последствий, при таких вариантах традиционно используются другие терапевтические подходы: системная химиотерапия и/или лучевая терапия.
Однако 5-летняя выживаемость без прогрессии при использовании стандартного режима химиотерапии карбоплатин/винкристин не превышает 45 %, что, естественно, не может считаться  Анна Киселеваудовлетворительным результатом. Повторные хирургические вмешательства и химиотерапия второй и последующих линий приводят к развитию тяжелых осложнений (утрата зрительных функций, развитие эндокринопатий, неврологического дефицита и др.). Применение лучевой терапии у пациентов с НЗГ позволяет в ряде случаев добиться контроля размеров опухоли, но сопряжено с высоким риском осложнений, особенно у детей младшей возрастной группы.
Анна Киселеваудовлетворительным результатом. Повторные хирургические вмешательства и химиотерапия второй и последующих линий приводят к развитию тяжелых осложнений (утрата зрительных функций, развитие эндокринопатий, неврологического дефицита и др.). Применение лучевой терапии у пациентов с НЗГ позволяет в ряде случаев добиться контроля размеров опухоли, но сопряжено с высоким риском осложнений, особенно у детей младшей возрастной группы.
 Таисия МихалевскаяТаким образом, поиск новых максимально безопасных и эффективных терапевтических опций в первую очередь необходим пациентам с инфантильными НЗГ срединной локализации (преимущественно хиазмально-селлярной области).
Таисия МихалевскаяТаким образом, поиск новых максимально безопасных и эффективных терапевтических опций в первую очередь необходим пациентам с инфантильными НЗГ срединной локализации (преимущественно хиазмально-селлярной области).
Особенности молекулярного патогенеза
Известно, что центральная роль в молекулярном патогенезе НЗГ принадлежит аберрантной активации сигнального пути MAPK, а основным механизмом запуска каскада является тандемная дупликация хромосомного региона 7q34, приводящая к формированию химерного онкогена KIAA1549: BRAF. Альтернативным механизмом активации сигнального пути MAPK являются активирующие мутации в киназном домене гена BRAF. Наиболее распространенный нуклеотидный вариант — миссенс-замена c.1799T>A, что в свою очередь влечет за собой замену аминокислотного остатка валина на глутаминовую кислоту в 600 кодоне протеинкиназы B-Raf — мутация BRAF V600E (см. рис. 1).
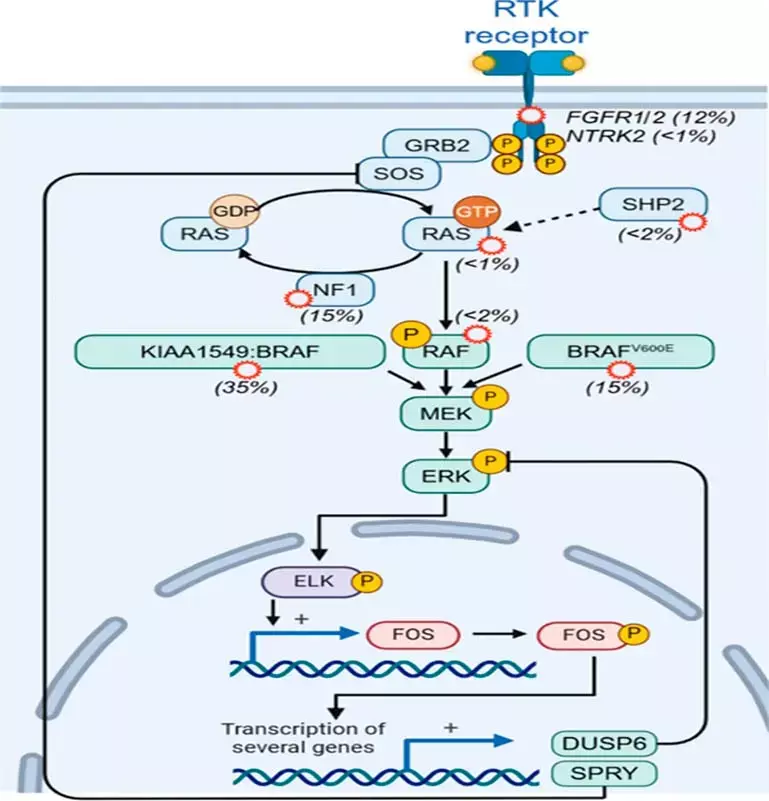
Рисунок 1. Сигнальные пути, вовлеченные в патогенез низкозлокачественных глиом.
Точечная мутация гена BRAF V600E при низкозлокачественных глиомах встречается с частотой 6–14 % и характеризуется неблагоприятным прогнозом при использовании только традиционных вариантов лечения (кривые выживаемости представлены на рис. 2). Частота встречаемости этой мутации варьирует в зависимости от гистологического варианта опухоли: 9 % при пилоцитарных астроцитомах, до 50 % при ганглиоглиомах и в пределах 70 % при плеоморфных ксантоастроцитомах.
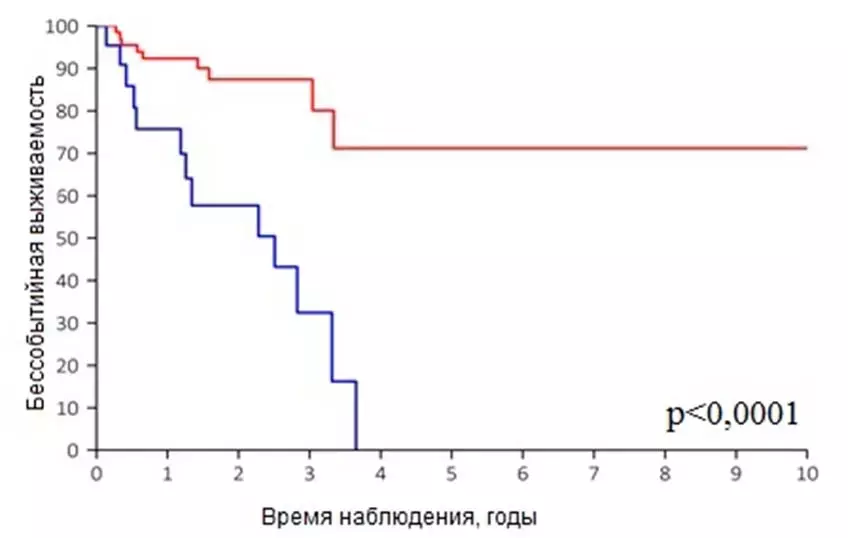
Рисунок 2. Бессобытийная выживаемость пациентов с пилоцитарными астроцитомами в зависимости от наличия BRAF V600E.
Наряду с мутацией BRAF V600E при НЗГ точками приложения терапевтического воздействия могут служить другие нуклеотидные варианты в компонентах сигнального пути RAS-RAF-MEK (FGFR1/2, MAP2K1), а также химерные транскрипты с участием генов RAF1, FGFR1,2, MYB, MYBL1, NTRK1,2,3, ALK, ROS1.
В отличие от НЗГ подавляющее большинство глиом высокой степени злокачественности имеют крайне неблагоприятный прогноз (выживаемость не превышает 15–25 % при применении максимально агрессивной мультимодальной терапии — хирургии, лучевой терапии, химиотерапии) и часто характеризуются отсутствием мишеней для таргетного воздействия. Исключение составляют лишь пациенты с инфантильными полушарными глиомами и злокачественными плеоморфными ксантоастроцитомами.
Инфантильная полушарная глиома — крайне редкая и малоизученная группа опухолей ЦНС с высокой частотой встречаемости перестроек генов ALK, ROS1 и NTRK1/2/3. Отличительной генетической особенностью плеоморфной ксантоастроцитомы 3-й степени злокачественности является мутация BRAF V600E, которая в ряде случаев сочетается с гомозиготной делецией CDKN2A/B.
Основными причинами недостаточной эффективности лучевой и химиотерапии является резистентность опухолей ЦНС к большинству цитостатиков, а также низкая радиочувствительность. Поскольку у большей части пациентов после нейрохирургического вмешательства определяется остаточная опухоль, крайне важной является разработка оптимальной стратегии их дальнейшего ведения.
Перспективные препараты
Наиболее перспективными таргетными препаратами для лечения НЗГ являются BRAF-ингибиторы у пациентов с точечной мутацией в гене BRAF V600E и MEK-ингибиторы при наличии химерного транскрипта KIAA1549:BRAF.
Препарат траметиниб является высокоселективным обратимым аллостерическим ингибитором МЕК1 и МЕК2, неконкурентным ингибитором АТФ. Положительный опыт лечения траметинибом НЗГ с наличием химерного транскрипта с участием гена BRAF был подтвержден в международных клинических исследованиях. Данный вид таргетной терапии активно используется в клинической практике РНПЦ ДОГИ.
Препарат дабрафениб является мощным селективным ингибитором BRAF-киназ с активирующими мутациями в кодоне 600. К настоящему моменту терапия BRAF-ингибиторами продемонстрировала многообещающие результаты, в особенности при опухолях ЦНС у детей и взрослых.
Лорлатиниб представляет собой селективный ингибитор рецепторов тирозинкиназы анапластической лимфомы и рецепторной тирозинкиназы ROS1 следующего поколения. Высокая интракраниальная активность лорлатиниба была многократно доказана в клинических исследованиях и на доклинических моделях, что с учетом высоких показателей эффективности и удовлетворительной переносимости дает основание для активного применения его для лечения детей с рефрактерными или рецидивирующими ROS1-/ALK-позитивными ВЗГ.
Также высокоселективной мишенью является наличие перестроек гена NTRK, которые встречаются при педиатрических ВЗГ в 5,3 % случаев. При NTRK-перестроенных опухолях ЦНС препаратами выбора в первой линии терапии рассматривается ларотректиниб, который является высокоселективным ингибитором тропомиозин-регулирующих киназ (TrkA/B/C) и показывает высокую эффективность у детей с опухолями ЦНС при наличии перестроек генов нейротрофической рецепторной тирозинкиназы (NTRK1/2/3). В 2018 году ларотректиниб стал первым TRK-ингибитором, одобренным FDA для терапии NTRK-перестроенных солидных опухолей, включая опухоли ЦНС для взрослых и детей.
Ниже представлены клинические случаи применения таргетной терапии на основании молекулярно-генетических маркеров опухоли в РНПЦ ДОГИ.
Клинические случаи
1. У девочки А. в возрасте 5 лет было выполнено тотальное удаление плеоморфной ксантоастроцитомы левой височной доли, Gr 2, BRAF+. В течение 6 лет наблюдения трижды отмечена прогрессия заболевания, в связи с чем проведены оперативные вмешательства с полным удалением новообразования.
На момент очередной прогрессии болезни помимо продолженного роста первичной опухоли выявлены метастазы по оболочкам головного мозга. Учитывая это, был назначен препарат дабрафениб в дозе 4,5 мг/кг/сутки внутрь. Через 6 месяцев приема препарата, по данным МРТ головного мозга, метастазы не определялись, объем опухоли уменьшился более чем в 2 раза, отмечена дальнейшая стабилизация заболевания в течение 3-х лет (см. рис. 3).
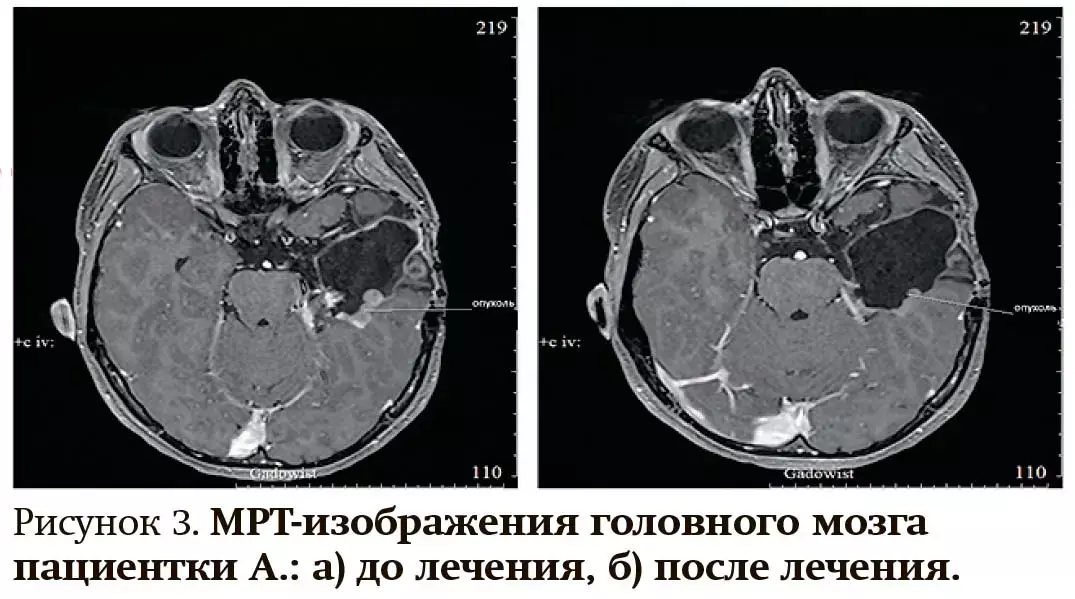
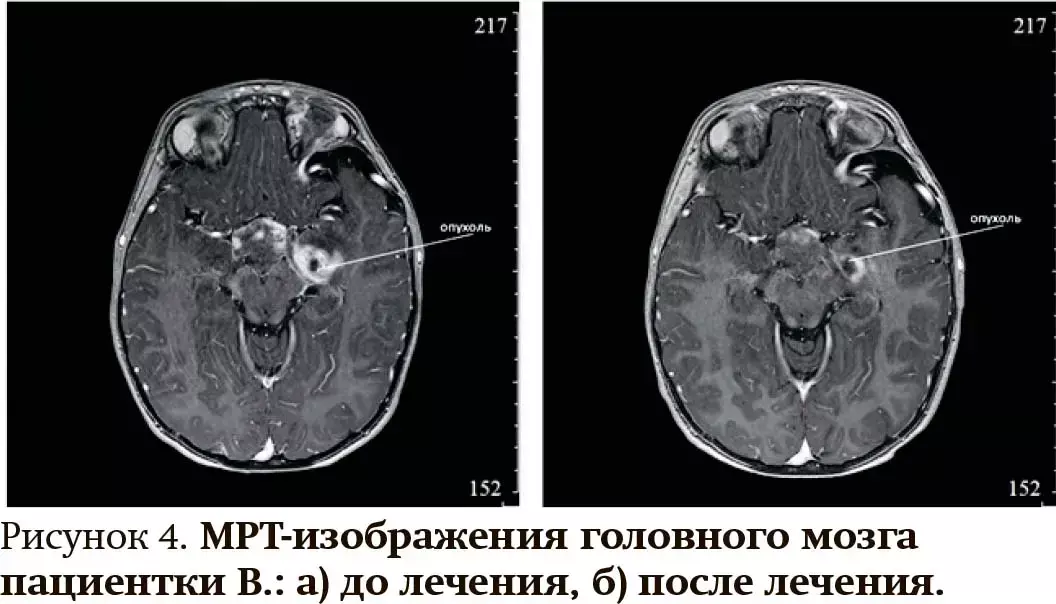
2. Девочке В. в возрасте 3 лет был выставлен диагноз «нейрофиброматоз 1-го типа, глиома оптико-хиазмальной области». Тактикой ведения пациентки было активное динамическое наблюдение врачом-онкологом с проведением МРТ головного мозга 2 раза в год. Через 2 года выявлен продолженный рост глиомы, в связи с чем назначен траметиниб в дозе 0,025 мг/кг 1 раз в сутки внутрь. Через 6 месяцев приема препарата диагностированы частичный ответ опухоли (уменьшение объема более чем на 25 %) и дальнейшая стабилизация опухолевого процесса в течение 5 лет (см. рис. 4).
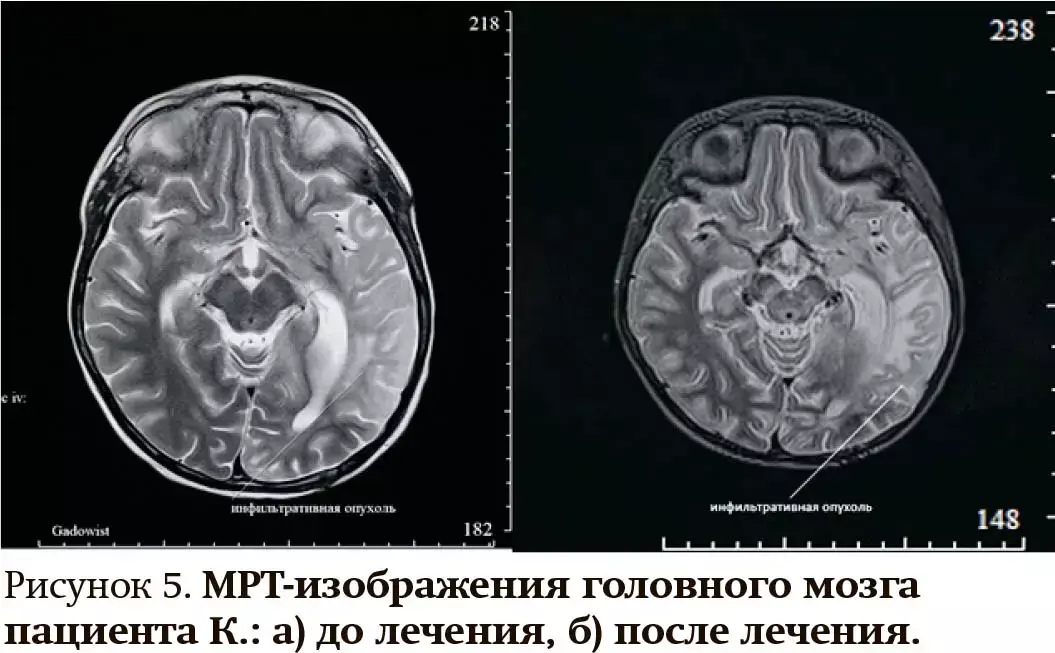
3. Мальчику К. в возрасте 5 лет выставлен диагноз «диффузная высокозлокачественная глиома педиатрического типа, IDH-wt, H3-wt с амплификацией гена EGFR левого полушария головного мозга, состояние после субтотального удаления образования». Проведена локальная лучевая терапия в СОД 54 Грей с последующим назначением таргетного препарата осимертиниб в дозе 80 мг 1 раз в сутки внутрь. Через год выявлена прогрессия заболевания, в связи этим выполнено повторное субтотальное удаление опухоли, локальная лучевая терапия в СОД 40 Грей с последующим назначением препарата в прежней дозе. В течение 6 месяцев наблюдается стабилизация заболевания (см. рис. 5).
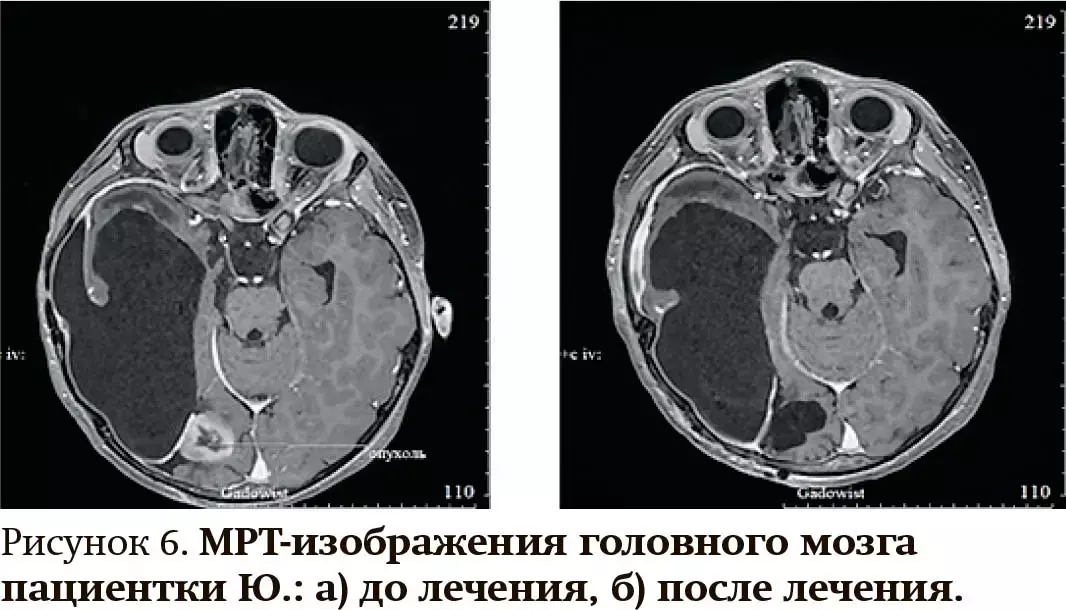
4. Девочке Ю. в возрасте 1 года было выполнено тотальное удаление инфантильной полушарной глиомы правой теменно-затылочной области, ассоциированной с реанжировкой ROS1, high grade. Через год наблюдения, по данным МРТ головного мозга, выявлен опухолевый узел в правой затылочной доле. Выполнено нейрохирургическое удаление образования и назначен препарат лорлатиниб в дозе 25 мг 1 раз в день внутрь. В течение 1 года и 6 месяцев наблюдается стабилизация заболевания (см. рис. 6).
Заключение
Современные подходы в лечении пациентов детского возраста с новообразованиями центральной нервной системы подразумевают определение индивидуальных молекулярно-генетических характеристик опухолевого процесса для подбора персонализированных вариантов терапии.
Применение таргетной терапии у детей с опухолями ЦНС в РНПЦ детской онкологии, гематологии и иммунологии позволило увеличить бессобытийную выживаемость пациентов с сохранением оптимального качества жизни.